Interferon-alpha/beta receptor
Interferon-alpha/beta receptor
Jump to navigation
Jump to search
| interferon (alpha, beta and omega) receptor 1 | |
|---|---|
| Identifiers | |
| Symbol | IFNAR1 |
| Alt. symbols | IFNAR |
| Entrez | 3454 |
| HUGO | 5432 |
| OMIM | 107450 |
| RefSeq | NM_000629 |
| UniProt | P17181 |
| Other data | |
| Locus | Chr. 21 q22.1 |
| interferon (alpha, beta and omega) receptor 2 | |
|---|---|
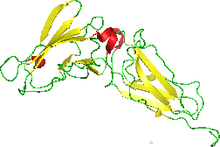 NMR structure of the interferon-binding ectodomain of the human interferon receptor.[1] | |
| Identifiers | |
| Symbol | IFNAR2 |
| Alt. symbols | IFNABR |
| Entrez | 3455 |
| HUGO | 5433 |
| OMIM | 602376 |
| RefSeq | NM_207585 |
| UniProt | P48551 |
| Other data | |
| Locus | Chr. 21 q22.1 |
The interferon-α/β receptor (IFNAR) is a virtually ubiquitous membrane receptor which binds endogenous type I interferon (IFN) cytokines. Endogenous human type I IFNs include many subtypes, such as interferons-α, -β, -ε, -κ, -ω, and -ζ.[2]
Contents
1 Function
2 Structure
2.1 Subunits
2.1.1 IFNAR1
2.1.2 IFNAR2
3 Signaling
3.1 Regulation
3.1.1 Differential expression
3.1.2 Endocytosis and downregulation
3.1.2.1 Clathrin-dependent endocytosis
3.1.3 Negative feedback mechanisms
4 Clinical implications
5 References
6 External links
Function[edit]
Activation of various innate immune signaling pathways (TLR3, TLR4, TLR7, TLR8, TLR9, cGAS, RIG-I, MDA-5) leads to the rapid induction of type I IFNs due to their (mostly) intronless gene structure.[3][4] The regulatory elements upstream of type I IFN genes differ, allowing differential transcription of type I IFNs in response to stimuli.[5] In particular, IFNβ contains a κB regulatory site, whereas IFNα subtypes do not. Production of specific type I IFNs is usually limited to a small number of type I IFN subtypes.[5] Once secreted, type I IFNs signal through IFNAR in a paracrine and autocrine manner.[6]
IFNAR is a heteromeric cell surface receptor composed of two subunits, referred to as the low affinity subunit, IFNAR1, and the high affinity subunit, IFNAR2.[7][8] Upon binding of type I interferons, IFNAR activates the JAK-STAT signalling pathway, along with MAPK, PI3K, and Akt signaling pathways.[2] IFNAR agonism results in transcriptional changes, with the potential to increase or suppress the transcription of over 2000 different genes.[9] For example, type I IFNs induce interferon-stimulated gene (ISG) expression, classically resulting in a robust anti-viral immune response. Additionally, IFNs largely impact cell health and viability, with effects on apoptosis, autophagy, cell differentiation, and proliferation.[10] The diverse effects of type I IFNs is likely dependent on the cellular and environmental context.[8]
Different responses, e.g. antiviral versus antiproliferative responses, to type I IFNs subtypes have been studied and proximal signaling, such as STAT phosphorylation, does not appear to correlate with the outcome. Furthermore, while differential effects manifest after several days of chronic stimulation, changes to receptor structure, orientation, or stoichiometry have not elucidated the cause for differential signaling via different type I IFN subtypes.[8][11] Current hypotheses for differential signaling include ligand-specific differences to the stability, or lifetime, of the ternary complex,[12] ligand-induced changes to internalization and trafficking of the receptor[8] and currently unappreciated differences to ligand-receptor structure.[12]
Structure[edit]
Type I IFN receptor forms a ternary complex, composed of its two subunits IFNAR1 and IFNAR2, and a type I IFN ligand. Ligand binding to either subunit is required for and precedes dimerization and activation of the receptor. Each subunit of IFNAR contains an N-terminal ligand binding domain (with two or four fibronectin type II-like subdomains, for IFNAR2 and IFNAR1, respectively), a transmembrane (TM) domain, and a cytoplasmic domain.[7] Each type I IFN ligand contains a "hotspot", or a sequence of conserved amino acids that are involved in binding to the receptor, specifically the high affinity receptor IFNAR2, which determines the affinity of each ligand for the receptor.[11]
Structural analysis of type I IFN receptor with different type I IFN ligand subtypes revealed a similar binding site for the different agonists.[8] Mutagenesis studies of type I IFNs, IFNAR1 and IFNAR2 demonstrated important binding residues, i.e. "hotspots", on the type I IFN subtypes which influenced its ability to bind to IFNAR2. Type I IFN binding to IFNAR1 was less strongly impacted by mutating single amino acids to alanine.[9] Importantly, structural studies have not revealed differences in ternary complex structures with IFNAR and various type I IFN subtypes, despite differences in ligand affinities.[8] The evolutionary conservation of type I IFN subtypes binding the same IFNAR receptor at the same site with differing affinities suggests that type I IFNs are nonredundant and potentially regulate different cellular responses.[2][11] Efforts to engineer a more potent IFNα2 elicited a cellular response similar to IFNβ, suggesting that the affinity of type I IFNs for IFNAR has an important role in regulating the downstream response.[9]
Subunits[edit]
Human IFNAR1 and IFNAR2 genes are located on chromosome 21q22.1.[13][14]
IFNAR1[edit]
IFNAR1 is the low affinity subunit, originally cloned in 1990, and is composed of four fibronectin type II-like (FNII-like) subdomains, termed SD1-4.[7][15] Type I IFNs bind SD1-3 with a typical binding affinity between 0.5–5μM; IFNα1 and IFNβ are exceptions with binding affinities of 220nM and 100nM, respectively.[7] Type I IFNs have a binding association rate of 5x105M/s with a variable dissociation rate that determines type I IFN subtype affinity for IFNAR1.[2]
IFNAR1 cytoplasmic domain associates with Tyk2 and Tyk2 is required for membrane expression of IFNAR1.[2] In the absence of Tyk2, a cytoplasmic IFNAR1 motif is phosphorylated, inducing receptor internalization.[9] IFNAR1 then localizes to the perinuclear endosomal compartment and is degraded.[8] Tyk2 null cells retain some responsiveness to IFNβ. However, responsiveness to IFNα is ablated, likely due to the reduced IFNAR1 membrane expression.[8] IFNAR1 binding to low affinity ligands, such as IFNα subtypes, has been proposed to be the rate limiting step in the ternary complex formation.[8] Therefore, if membrane levels of IFNAR1 are too low, the binary IFNα-IFNAR2 complex will be unable to recruit it to induce signaling. This hypothesis is supported by observations that cell lines with low IFNAR1 expression respond to IFNβ but not IFNα.[8]
IFNAR2[edit]
IFNAR2 is the high affinity subunit, originally cloned in 1994, composed of two FNII-like subdomains, termed D1 and D2.[7][15] Type I IFNs bind D1 and D2 with a typical binding affinity between 0.4–5nM; IFNβ binds at a slightly lower affinity (0.1nM).[7] Type I IFNs have a binding association rate of 10−6–10−7M/s with a variable dissociation rate that determines type I IFN subtype affinity for IFNAR2.[7]
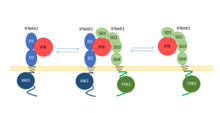
This modified schematic shows each subunit independently binding the type I IFN ligand and recruiting the corresponding subunit.[9] The IFNAR1 and IFNAR2 binding motifs on type I IFNs are approximately 180 degrees from each other.[9] IFNAR is composed of two subunits, IFNAR1 and IFNAR2. Each IFNAR subunit contains an N-terminal ligand binding domain, a transmembrane domain, and a C-terminal intracellular tail.[9] IFNAR1 is the low affinity subunit, with four N-terminal subdomains. Binding to type I IFN is mediated by subdomains SD1, SD2 and SD3. IFNAR1's C-terminal tail interacts with Tyk2.[7] IFNAR2 is the high affinity subunit, composed of two N-terminal subdomains. Binding to type I IFN is mediated by D1 and D2. IFNAR2's C-terminal tail interacts with JAK1.[7]
IFNAR2 is expressed as three isoforms due to alternative splicing, exon skipping, and multiple polyadenylation sites; IFNAR2a (a soluble form lacking the TM domain), IFNAR2b (a soluble form lacking the cytoplasmic domain), and IFNAR2c (the membrane bound, signaling-competent form).[2] The cytoplasmic domain of IFNAR2c associates with JAK1.[2]
Signaling[edit]
Type I IFNs bind to IFNAR1 or IFNAR2, forming a binary complex.[9] The binary complex further recruits the remaining IFNAR subunit, completing the ternary complex and activating downstream JAK/STAT signaling.[9] IFN ligation to IFNAR brings the receptor associated kinases, JAK1 and Tyk2, into close proximity, resulting in kinase transphosphorylation and subsequent phosphorylation of tyrosines on IFNAR1 and IFNAR2.[5][7] Phosphotyrosine residues on IFNAR1 and IFNAR2 recruit STAT proteins[7] (classically STAT1, STAT2, or STAT3, although STAT4, STAT5, and STAT6 may play a role in certain cell types[16]) via their SH2 domains. Once recruited, STAT proteins are phosphorylated by which induces their homo- or heterodimerization.[17] These dimers translocate to the nucleus, binding interferon-stimulated response elements (ISRE) and gamma activating sequences (GAS), promoting gene transcription.[7][17]
Regulation[edit]
Under normal conditions, type I IFN levels are homeostatically regulated to balance the benefits of protection from viral infection with the other cellular effects associated with type I IFN signaling, such as apoptosis, cell cycle arrest, and immune modulation.[9] The stringent regulation of type I IFN signaling suggests the importance of timing and location.[5] Type I IFN signaling is controlled through various mechanisms, including differential expression of signaling components,[18] differential signaling following IFNAR engagement,[9] endocytosis and downregulation of the receptors.[2][9][15] and negative feedback mechanisms[9]
Differential expression[edit]
Responsiveness to type I IFNs requires the expression of signaling components within a given target cell, including IFNAR and STATs. Expression levels and post-translational modifications to IFNAR and STATs can modify cell responsiveness.[18] Importantly, signaling is impacted by different STAT protein expression levels and activation of STAT heterodimers and STAT homodimers; reducing STAT expression levels alters antiviral, antiproliferative and inflammatory responses to type I IFNs.[18] Conversely, increased expression of STAT1 and IRF9 maintain ISG expression, even in the absence of on-going cytokine receptor signaling, thereby amplifying IFNAR signaling.[18] Because type I IFN signaling modulates proteins involved in protein translation, it can also indirectly alter protein levels of induced genes and proteins involved in pathway signaling.[18]
Type I IFN subtypes, ranging from 30–70% homology, all bind the same receptor.[9] Biased agonism has been studied in order to better understand how a single receptor responds differently to multiple cognate ligands, culminating in disparate outcomes.
Endocytosis and downregulation[edit]
IFNAR1 and IFNAR2 can be internalized through endocytosis in response to agonism through clathrin-dependent and clathrin-independent mechanisms.[2] IFNAR subunits can be differentially downregulated following IFN stimulation. For example, membrane IFNAR1 is reduced in response to IFNα, but surface levels IFNAR1 and IFNAR2 are downregulated in response to IFNβ binding.[8] In agreement with these observations, IFNAR internalization is often associated with the respective agonist's ability to induce an anti-proliferative effect.[7]
IFNAR subunit internalization is also observed under basal conditions, with greater basal turnover of IFNAR1 than IFNAR2.[2] IFNAR1 contains a degron, or a motif for ubiquitination, which allows for its internalization and degradation.[2] Tyk2 may block this degron, preventing its internalization.[8]
Clathrin-dependent endocytosis[edit]
Following receptor agonism, the C-terminus of IFNAR is phosphorylated, followed by its ubiquitination and internalization.[8][15] IFNAR surface expression is maintained in the presence of stimulation when the clathrin-dependent endocytosis pathway is inhibited with siRNA knockdown of clathrin or using a small molecule inhibitor of the GTPase dynamin.[15] Internalization can result in degradation of the receptor, thereby reducing membrane expression,[15] or it can result in recycling of the receptor without an extended impact on membrane receptor levels.[2] However, clathrin-mediated endocytosis may also serve to concentrate the IFNAR receptors and signaling components, thereby amplifying signaling.[15] Electron microscopy experiments show IFNAR receptors concentrated in clathrin-coated pits, and inhibition of clathrin-mediated endocytosis resulted in reduced phosphorylation of JAK1, Tyk2, STATs and reduced STAT nuclear translocation.[9][15]
Negative feedback mechanisms[edit]
Negative regulators of type I IFN signaling, such as suppressor of cytokine signaling 1 (SOCS1) and ubiquitin-specific peptidase 18 (USP18), are induced after 4 hours.[9] SOCS1 inhibits type I IFN signaling by inhibiting JAKs and binding phosphorylated Tyk2.[9] USP18 binds to the C terminus of IFNAR2.[9] USP18 may regulate type I IFN signaling by interfering with ternary complex formation, not through its peptidase function.[7] Loss of USP18 results in an inflammatory interferon-mediated CNS disease.[3]
Clinical implications[edit]
Type I IFN can provide both beneficial or deleterious effects in a variety of diseases. Type I IFN is thought to be a driver in multiple autoimmune diseases and may have a role in chronic infection.[18] Conversely, type I IFNs are also prescribed as therapeutics in other disease indications.[18]
In particular, type I IFN are implicated in the pathogenesis of the in the following autoimmune diseases: systemic lupus erythematosus (SLE), Sjogren's syndrome, systemic sclerosis, rheumatoid arthritis (RA), and myositis.[18] Increased levels of intracerebral IFNα are also thought to play a detrimental role in Aicardi–Goutières syndrome (AGS), HIV-associated dementia and CNS lupus.[12] While type I IFNs are one of the classical cytokines required for an effect antiviral response, higher type I IFN levels are associated with worsening disease in bacterial infections, such as tuberculosis and lepromatous leprosy.[18] Type I IFN is also being investigated for a potential role in neurodegeneration; loss of IFNAR expression prolonged survival in murine models of amyotrophic lateral sclerosis (ALS).[12]
IFNα has also been used in the clinic for the treatment of type I IFN responsive hematological malignancies, such as chronic myelogenous leukemia (CML), multiple myeloma, and hairy cell leukemia.[16] Importantly, type I IFN-resistant cancers have been shown to have low or absent IFNAR expression.[16] Additionally, IFNα was also given therapeutically for the treatment of some potentially chronic viral infections, such as hepatitis B and hepatitis C. Paradoxically, IFNβ was first-line treatment for the central autoimmune disease, multiple sclerosis (MS), although the mechanism of action for IFNβ in MS has not been definitively demonstrated.[12][16][19]
References[edit]
^ PDB: 1n6u; Chill JH, Quadt SR, Levy R, Schreiber G, Anglister J (July 2003). "The human type I interferon receptor: NMR structure reveals the molecular basis of ligand binding". Structure. 11 (7): 791–802. doi:10.1016/S0969-2126(03)00120-5. PMID 12842042..mw-parser-output cite.citationfont-style:inherit.mw-parser-output qquotes:"""""""'""'".mw-parser-output code.cs1-codecolor:inherit;background:inherit;border:inherit;padding:inherit.mw-parser-output .cs1-lock-free abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/6/65/Lock-green.svg/9px-Lock-green.svg.png")no-repeat;background-position:right .1em center.mw-parser-output .cs1-lock-limited a,.mw-parser-output .cs1-lock-registration abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/d/d6/Lock-gray-alt-2.svg/9px-Lock-gray-alt-2.svg.png")no-repeat;background-position:right .1em center.mw-parser-output .cs1-lock-subscription abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/a/aa/Lock-red-alt-2.svg/9px-Lock-red-alt-2.svg.png")no-repeat;background-position:right .1em center.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registrationcolor:#555.mw-parser-output .cs1-subscription span,.mw-parser-output .cs1-registration spanborder-bottom:1px dotted;cursor:help.mw-parser-output .cs1-hidden-errordisplay:none;font-size:100%.mw-parser-output .cs1-visible-errorfont-size:100%.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration,.mw-parser-output .cs1-formatfont-size:95%.mw-parser-output .cs1-kern-left,.mw-parser-output .cs1-kern-wl-leftpadding-left:0.2em.mw-parser-output .cs1-kern-right,.mw-parser-output .cs1-kern-wl-rightpadding-right:0.2em
^ abcdefghijkl de Weerd NA, Nguyen T (May 2012). "The interferons and their receptors—distribution and regulation". Immunology and Cell Biology. 90 (5): 483–91. doi:10.1038/icb.2012.9. PMID 22410872.
^ ab McGlasson S, Jury A, Jackson A, Hunt D (September 2015). "Type I interferon dysregulation and neurological disease". Nature Reviews. Neurology. 11 (9): 515–23. doi:10.1038/nrneurol.2015.143. PMID 26303851.
^ Nallar SC, Kalvakolanu DV (August 2014). "Interferons, signal transduction pathways, and the central nervous system". Journal of Interferon & Cytokine Research. 34 (8): 559–76. doi:10.1089/jir.2014.0021. PMC 4118707. PMID 25084173.
^ abcd Hertzog PJ, Williams BR (June 2013). "Fine tuning type I interferon responses". Cytokine & Growth Factor Reviews. 24 (3): 217–25. doi:10.1016/j.cytogfr.2013.04.002. PMID 23711406.
^ Coccia EM, Battistini A (March 2015). "Early IFN type I response: Learning from microbial evasion strategies". Seminars in Immunology. 27 (2): 85–101. doi:10.1016/j.smim.2015.03.005. PMID 25869307.
^ abcdefghijklmn Piehler J, Thomas C, Garcia KC, Schreiber G (November 2012). "Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation". Immunological Reviews. 250 (1): 317–34. doi:10.1111/imr.12001. PMC 3986811. PMID 23046138.
^ abcdefghijklm Uzé G, Schreiber G, Piehler J, Pellegrini S (2007). "The receptor of the type I interferon family". Current Topics in Microbiology and Immunology. Current Topics in Microbiology and Immunology. 316: 71–95. doi:10.1007/978-3-540-71329-6_5. ISBN 978-3-540-71328-9. PMID 17969444.
^ abcdefghijklmnopqr Schreiber G, Piehler J (March 2015). "The molecular basis for functional plasticity in type I interferon signaling". Trends in Immunology. 36 (3): 139–49. doi:10.1016/j.it.2015.01.002. PMID 25687684.
^ Trinchieri G (September 2010). "Type I interferon: friend or foe?". The Journal of Experimental Medicine. 207 (10): 2053–63. doi:10.1084/jem.20101664. PMC 2947062. PMID 20837696.
^ abc Ng CT, Mendoza JL, Garcia KC, Oldstone MB (January 2016). "Alpha and Beta Type 1 Interferon Signaling: Passage for Diverse Biologic Outcomes". Cell. 164 (3): 349–52. doi:10.1016/j.cell.2015.12.027. PMC 4733246. PMID 26824652.
^ abcde Owens T, Khorooshi R, Wlodarczyk A, Asgari N (March 2014). "Interferons in the central nervous system: a few instruments play many tunes". Glia. 62 (3): 339–55. doi:10.1002/glia.22608. PMID 24588027.
^ Lutfalla G, Roeckel N, Mogensen KE, Mattei MG, Uzé G (October 1990). "Assignment of the human interferon-α receptor gene to chromosome 21q22.1 by in situ hybridization". Journal of Interferon Research. 10 (5): 515–7. doi:10.1089/jir.1990.10.515. PMID 2148760.
^ Lutfalla G, Holland SJ, Cinato E, Monneron D, Reboul J, Rogers NC, Smith JM, Stark GR, Gardiner K, Mogensen KE (October 1995). "Mutant U5A cells are complemented by an interferon-αβ receptor subunit generated by alternative processing of a new member of a cytokine receptor gene cluster". The EMBO Journal. 14 (20): 5100–8. PMC 394613. PMID 7588638.
^ abcdefgh Claudinon J, Monier MN, Lamaze C (2007). "Interfering with interferon receptor sorting and trafficking: impact on signaling". Biochimie. 89 (6–7): 735–43. doi:10.1016/j.biochi.2007.03.014. PMID 17493737.
^ abcd Deonarain R, Chan DC, Platanias LC, Fish EN (2002). "Interferon-α / β-receptor interactions: A complex story unfolding". Current Pharmaceutical Design. 8 (24): 2131–7. doi:10.2174/1381612023393288. PMID 12369858.
^ ab Hebenstreit D, Horejs-Hoeck J, Duschl A (May 2005). "JAK/STAT-dependent gene regulation by cytokines". Drug News & Perspectives. 18 (4): 243–9. doi:10.1358/dnp.2005.18.4.908658. PMID 16034480.
^ abcdefghi Ivashkiv LB, Donlin LT (January 2014). "Regulation of type I interferon responses". Nature Reviews. Immunology. 14 (1): 36–49. doi:10.1038/nri3581. PMC 4084561. PMID 24362405.
^ Kieseier BC (June 2011). "The mechanism of action of interferon-β in relapsing multiple sclerosis". CNS Drugs. 25 (6): 491–502. doi:10.2165/11591110-000000000-00000. PMID 21649449.
External links[edit]
Receptor,+Interferon+alpha-beta at the US National Library of Medicine Medical Subject Headings (MeSH)
Categories:
- Genes on human chromosome 21
- Type II cytokine receptors
(window.RLQ=window.RLQ||).push(function()mw.config.set("wgPageParseReport":"limitreport":"cputime":"0.640","walltime":"0.837","ppvisitednodes":"value":2525,"limit":1000000,"ppgeneratednodes":"value":0,"limit":1500000,"postexpandincludesize":"value":168554,"limit":2097152,"templateargumentsize":"value":1134,"limit":2097152,"expansiondepth":"value":12,"limit":40,"expensivefunctioncount":"value":4,"limit":500,"unstrip-depth":"value":1,"limit":20,"unstrip-size":"value":73796,"limit":5000000,"entityaccesscount":"value":2,"limit":400,"timingprofile":["100.00% 673.506 1 -total"," 62.22% 419.084 1 Template:Reflist"," 45.99% 309.757 19 Template:Cite_journal"," 13.03% 87.750 2 Template:Infobox_protein"," 9.00% 60.634 2 Template:Infobox"," 8.81% 59.353 10 Template:Navbox"," 6.99% 47.082 1 Template:Use_dmy_dates"," 4.79% 32.235 2 Template:DMCA"," 4.32% 29.079 2 Template:Dated_maintenance_category"," 3.61% 24.293 1 Template:Cytokine_receptors"],"scribunto":"limitreport-timeusage":"value":"0.310","limit":"10.000","limitreport-memusage":"value":4461557,"limit":52428800,"cachereport":"origin":"mw2231","timestamp":"20180929142557","ttl":1900800,"transientcontent":false);mw.config.set("wgBackendResponseTime":90,"wgHostname":"mw2163"););

 Clash Royale CLAN TAG
Clash Royale CLAN TAG